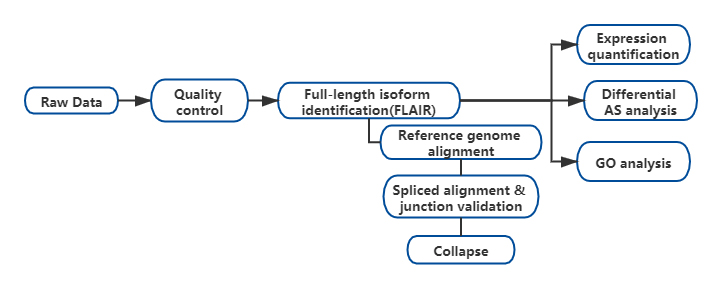
Application of Nanopore full-length transcriptome sequencing in isoform-level AS study
TRANSCRIPTOMICS
nature
COMMUNICATIONS
Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns
Full-length transcripts| Nanopore sequencing| Alternative isoform analysis
Background
Somatic mutations in splicing factor SF3B1 has been widely reported to associate with various cancers, including chronic lymphocytic leukemia(CLL), uveal melanoma, breast cancer, etc. In addition, short-read transcriptomic studies have revealed aberrant splicing patterns induced by SF3B1 mutations. However, studies on these alternative splicing patterns has long been limited to event-level and lack of knowledge on isoform-level due to the limitation of short-read assembled transcripts. Here, nanopore sequencing platform was introduced to generate full-length transcripts, which empowered inverstigation on AS isoforms.
Experimental Design
Experiments
Bioinformatic Analysis
Results
A total of 257 million reads were generated from 6 CLL samples and 3 B-cells. On average of 30.5% of these reads were identified as full-length transcripts.
FLAIR Identified 326,699 high-confidence spliced isoforms, 90% of which are novel isoforms. Most of these unannotated isoforms were found to be novel combinations of known splice juntions(142,971), while the rest novel isoforms contained either retained intron(21,700) or novel exon(3594).
Long-read sequences empowers identification of mutant SF3B1-K700E -altered splice sites at isoform-level. 35 alternative 3’SSs and 10 alternative 5’SSs were found to be significantly differentially spliced between SF3B1-K700E and SF3B1-WT. 33 of the 35 alterations were newly discovered by long-read sequences. In Nanopore data, the distribution of distance between SF3B1-K700E-altered 3’SSs to canonical sites peaks is around -20 bp, which is significantly differed from a control distribution, similar to what has been reported in CLL short-read sequences. Isoforms of ERGIC3 gene were analyzed, where a novel isoform containing the proximal splice site were found more abundant in SF3B1-K700E . Both proximal and distal 3’SS were associated with distinced AS patterns generating multiple isoforms.
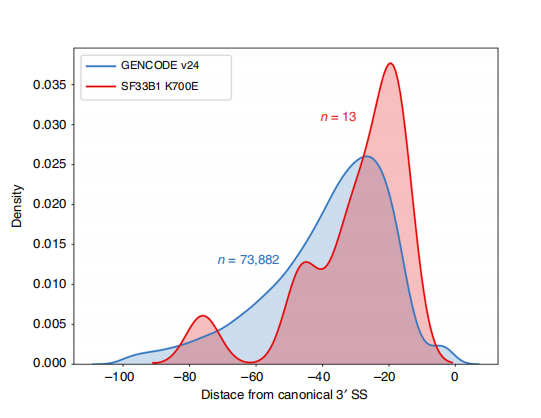
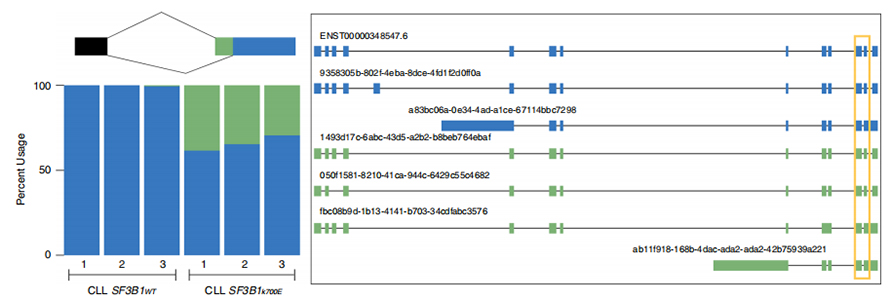
Figure 2. Alternative 3′ splicing patterns identified with nanopore sequencing data
IR event usage analysis has been long limited in short-read based analysis due to confidence in IR identification and quantification. Expression of IR isoforms in SF3B1-K700E and SF3B1-WT were quantified based on nanopore sequences, revealing a global down-regulation of IR isoforms in SF3B1-K700E .
Figure 4. Agriculture intensity and network connectivity across three farming systems (A and B); Random forest analysis(C) and Relationship between agricultural intensity and AMF colonization (D)
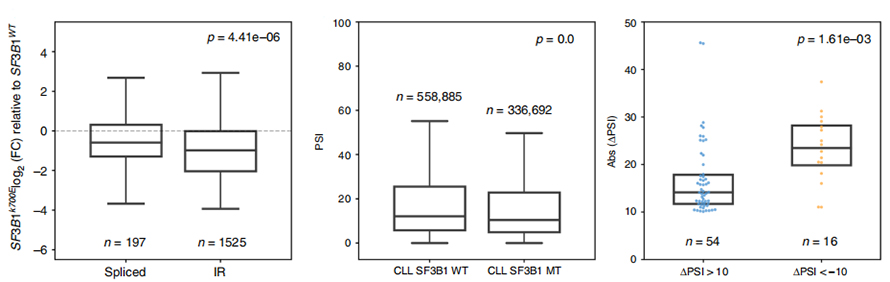
Figure 3. Intron rentention events are more strongly downregulated in CLL SF3B1-K700E
Technology
Nanopore Long-read Sequencing
Performance of full-length transcriptome sequencing
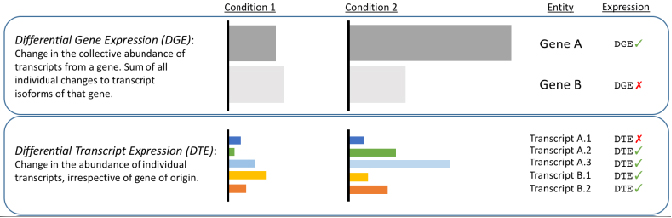
√ Transcript-level differential analysis -Reveal changes hiden by short-reads
Reference
Tang A D , Soulette C M , Baren M J V , et al. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns[J]. Nature Communications.
Tech and Highlights aims at sharing most recent successful application of different high-throughput sequencing technologies in various reseach arena as well as brilliant ideas in experimental design and data mining .